- CHROMATOGRAPHIE
- CHROMATOGRAPHIELa chromatographie est une méthode d’analyse chimique consistant à séparer les constituants d’un mélange. Aussi est-elle universellement employée, au laboratoire comme dans l’industrie, tant pour l’analyse proprement dite que pour la séparation ou l’isolement des corps purs.Sa découverte, qui remonte à 1903, est due au botaniste russe Tswett. Il avait observé la séparation des constituants de la chlorophylle brute lorsque sa solution montait, par capillarité, le long d’un papier filtre. Dans ces conditions, en effet, des zones colorées se forment, qui correspondent à la chlorophylle (verte), à la xantophylle (jaune) et au carotène (rouge).Redécouverte après une longue période d’oubli, entre 1930 et 1940, la chromatographie s’est généralisée à l’analyse de substances incolores – allant des gaz aux composés macromoléculaires –, organiques ou minérales; son nom, dès lors, ne se justifie guère. Le schéma initial est très peu modifié dans la chromatographie sur papier, particulièrement utile en biochimie, et permettant l’étude d’échantillons de quelques microgrammes. Il y a, dans ce cas, une succession d’échanges, les processus élémentaires, entre la solution et le papier, qu’on retrouve dans toutes les autres techniques chromatographiques sous forme d’échange entre une phase (ou milieu homogène) mobile et une phase fixe. Cela n’est pas sans analogie avec la distillation, où ces échanges ont lieu entre un liquide et un gaz, tous deux mobiles et circulant à contre-courant dans une colonne. Mais, alors que dans une excellente colonne à distiller ces processus élémentaires ne dépassent guère la centaine (on dit qu’elle a une centaine de «plateaux théoriques»), ils peuvent être plusieurs milliers en chromatographie en phase gazeuse, voire des centaines de milliers, lorsqu’on procède avec les colonnes capillaires.Quelle que soit la technique considérée, la chromatographie a donc une efficacité qui dépasse de très loin celle de tous les autres procédés de fractionnement. Les produits à séparer le seront, pourvu qu’ils aient des propriétés différentes vis-à-vis des deux phases en présence: la plus infime différence est amplifiée par la répétition d’un nombre très important d’échanges. Il faut joindre à cette qualité la possibilité de travailler sur les composés les plus fragiles qui peuvent être traités en solution à la température ambiante. La chromatographie, technique analytique à l’origine, est devenue le procédé de préparation le plus pratique pour des substances aussi diverses que les acides aminés ou les terres rares, difficiles à isoler des mélanges qui en contiennent plusieurs homologues. Cependant, si elle est dès l’origine très facilement adaptée à la micro- et ultramicro-analyse, son extension à des préparations industrielles est plus récente.1. Bases de la chromatographieThéorie de l’analyse chromatographiqueLe principe d’une séparation chromatographique schématisée à la figure 1, dans le cas d’un mélange de deux constituants A et B, est généralisable sans peine à un nombre plus grand de systèmes plus complexes.Le mélange A + B, dissous dans un solvant S, est introduit au sommet d’une colonne contenant la phase fixe. La quantité de A + B doit être très faible par rapport à ce que peut retenir la totalité de la phase stationnaire. Les deux substances se fixent alors par échange en haut de la colonne qui est saturée sur une très faible longueur par le mélange. Une zone, ayant les mêmes proportions de A et B que la solution, se forme ainsi.Par la suite, la colonne sera soumise à une percolation par un solvant qui peut être identique à S ou en différer, et qui peut, d’ailleurs, voir sa composition modifiée au cours de l’opération. Cette étape s’appelle élution , et elle détermine une migration de la zone vers le bas de la colonne. Le solvant frais (ou éluant) dissout, en effet, progressivement les substances fixées sur la colonne pour les abandonner un peu plus loin sur les portions vierges de celle-ci. Ces échanges se passent au niveau de chaque grain de phase stationnaire et sont régis par les coefficients de partage de A et B. Ceux-ci n’ayant aucune raison d’être identiques, la vitesse de migration de la zone est différente pour ces deux composants. La colonne se charge progressivement en A vers le bas, si c’est A qui a le coefficient de partage le plus favorable à la dissolution dans le liquide mobile, et en B vers l’arrière. Bientôt la zone se dédouble et la séparation est effective entre A et B.Une technique, parfois utilisée au niveau de la préparation, consiste à isoler effectivement, en les sortant de la colonne, les sections contenant les produits séparés, et à extraire chacun d’eux par un solvant convenable. Mais dans la majorité des cas, on procède au développement par élution. On fait passer une quantité suffisante d’éluant pour que le contenu des zones A et B, lorsque celles-ci sont parvenues au bout de la colonne, passent successivement en solution, et y soit détecté, puis éventuellement récupéré.On comprend que, dans cette opération, le processus élémentaire s’effectue sur une longueur très faible, de l’ordre de grandeur des grains de phase stationnaire et que le nombre de «plateaux théoriques» soit très élevé. Les signaux obtenus à la suite d’une mesure par un détecteur, fournissent un enregistrement graphique de la concentration en fonction du temps t ou du volume V élué, dont la forme est celle d’une courbe de Gauss. On définit, comme en distillation, la longueur équivalente à un plateau théorique: celle d’une colonne suffisante pour que la phase mobile en sorte en équilibre avec la phase fixe. D’un ordre de grandeur comparable à celui de quelques grains de phase fixe, c’est une valeur expérimentale, et l’on démontre que le nombre N de plateaux théoriques est: N = 16 (V2R/Wb size=1 2 si VR est le volume d’élution d’un produit, Wb la largeur à la base du pic de la courbe enregistrée dans le même système de mesure.Pour une colonne donnée, ce nombre varie avec les conditions expérimentales; l’efficacité de l’analyse en dépend. N augmente avec la longueur de la colonne; mais le calcul montre que, si l’écart entre deux zones est bien proportionnel à la distance qu’elles ont parcourue, leur largeur s’accroît comme sa racine carrée: néanmoins, la concentration des produits recherchés dans l’éluant peut devenir trop faible pour qu’ils soient récupérables, ou seulement décelables. La chromatographie rencontre là ses limites.2. Chromatographie en phase liquideLa chromatographie en phase liquide (CPL), un moment éclipsée par la chromatographie en phase gazeuse (CPG), a fait des progrès considérables grâce à la miniaturisation des colonnes et à la détection en continu des fractions. L’appareil est constitué d’une colonne en acier inoxydable d’une longueur de l’ordre de 200 mm et de diamètre intérieur de 4 mm, remplie de particules de taille inférieure à 20 猪m; cette colonne est précédée d’une pompe à haute pression, permettant d’injecter le solvant sous une pression de plusieurs dizaines de bars. Des microvannes assurent l’introduction de quelques microlitres de solution à analyser, en tête de colonne. La détection des fractions est le plus souvent faite par absorption UV ou réfractométrie; elle est suivie de l’enregistrement graphique des signaux ou de leur traitement direct par calculateur. Toutes les opérations peuvent être automatisées dans le cas d’analyses de routine. Les quantités analysées sont de l’ordre du milligramme et l’efficacité varie entre 1 000 et 10 000 plateaux théoriques; sur des colonnes appropriées on sépare par exemple tous les amino-acides naturels, alors des quantités de substance aussi faibles que 10-12 g peuvent être décelées.Par ailleurs, l’extension de la CPL au niveau de la préparation est facile pour des quantités de quelques grammes, et le laboratoire l’emploie très couramment. Le problème à l’échelle industrielle est de trouver des phases fixes dont le prix ne soit pas prohibitif; la CPL est ainsi compétitive pour des produits comme les antibiotiques ou les protéines. (Les premières unités, capables de produire quelques centaines de kilogrammes par jour, fonctionnent.)Les méthodes utilisables en CPL sont très nombreuses, et un choix judicieux fournira la solution de n’importe quel problème analytique.Chromatographie par adsorptionLes phases fixes adsorbantes les plus employées sont l’alumine et le gel de silice, à des granulométries, porosités et activités adaptées au cas à résoudre. Quand on met un solide en présence d’un gaz ou d’un liquide, il fixe sur sa surface les molécules de la phase fluide, qui s’y trouvent relativement concentrées: elles y sont adsorbées. Mais ce phénomène est plus ou moins marqué suivant la nature des surfaces, suivant celle des corps en présence, et dépend de la température. Pour un adsorbant donné, l’adsorption d’un produit dissous dans un liquide solvant peut être caractérisée par le rapport x/m de la masse de ce produit adsorbée, x , à celle, m , du solide. Ce rapport dépend de la température et de la concentration c , et la courbe obtenue en portant x/m en fonction de c à température constante est une isotherme d’adsorption. Ces courbes ne coïncident pas pour deux solutés (corps dissous) différents et, si leur concentration est égale, l’un d’eux est plus fortement adsorbé par le solide. Plus généralement, chaque fois que l’on met en contact une solution avec un solide, on observe une diminution de la concentration de chacun de ses éléments dans un rapport différent, tandis qu’il s’en fixe sur le solide une quantité dépendant de ses caractéristiques d’adsorption. Les rapports des concentrations des solutés sur le solide sont, par suite, différents de ce qu’ils sont dans la solution.Réciproquement, en plaçant au contact d’un solvant pur un solide qui supporte des composés adsorbés, chacun de ceux-ci se dissoudra plus ou moins suivant sa solubilité et son affinité pour le solide jusqu’à ce que l’équilibre s’établisse entre les deux phases.Un cycle d’adsorption-désorption constitue le processus élémentaire de la chromatographie par adsorption; il se répète en principe de grain en grain à mesure que le fluide mobile progresse le long du solide.Chromatographie de partage liquide-liquideAu lieu d’effectuer des échanges de solutés entre un liquide (ou un gaz) et un solide, on peut le faire entre deux liquides non miscibles. On sait qu’il existe alors pour chaque corps dissous un rapport constant, appelé coefficient de partage, entre ses concentrations dans chacun des liquides: c /c = K. K est caractéristique d’une espèce chimique pour le couple de solvants L et L . L’isotherme de partage est une droite, ce qui entraîne une simplification par rapport aux phénomènes d’adsorption où les isothermes s’écartent notablement de cette forme. La difficulté pour utiliser le partage entre deux liquides en chromatographie est de fractionner l’un des liquides en nombreux petits domaines immobiles, analogues aux grains d’adsorbant. On peut la résoudre en imprégnant du liquide qui doit former la phase stationnaire un solide poreux de fine granulométrie. Par exemple, les grains de gel de silice, qui sont hydratés, constituent une phase stationnaire aqueuse dont les seules propriétés qui entrent en ligne de compte sont celles de l’eau. Très souvent, en effet, le rôle d’adsorbant de la silice est accessoire, et la chromatographie est fondée sur l’échange entre le solvant et une phase imprégnée d’eau soit, pour l’essentiel des 漣 OH liés aux Si. De là, on en est venu au greffage chimique d’autres groupements sur la silice, par exemple des fonctions 漣 NH2. La phase fixe est polaire et hydrophile; les composés à séparer se partageront entre elle et le solvant peu ou pas polaire (par exemple un hydrocarbure), selon leur caractère plus ou moins hydrophile. On dit qu’il s’agit d’une CPL en phases normales.Mais elle est largement supplantée par la chromatographie en phases inversées où le support solide est greffé par des groupes chimiques comme des chaînes hydrocarbonées en C18, des noyaux aromatiques ou autres fonctions non polaires. Le partage est alors gouverné par les interactions hydrophobes des solutés avec la phase fixe, alors que le solvant est polaire (par exemple alcool hydraté). Le succès de cette technique tient au très grand choix de phases inversées proposées par les fabricants. L’élution, avec plusieurs solvants dont on fait varier les concentrations relatives suivant un programme, en facilite l’optimisation.Les colonnes à phases inversées conviennent à l’analyse de toutes les substances organiques sous réserve de modifications convenables dans certains cas particuliers. C’est ainsi que les composés ionisables, comme les acides organiques, se séparent mal, donnant des pics dédoublés (la molécule et l’ion). L’addition au solvant d’acide acétique supprime l’ionisation des solutés qui donnent alors des pics normaux. Pour les composés très ionisés comme des sels d’acides sulfoniques, on fait une chromatographie par appariement d’ions : on ajoute un ion à longue chaîne hydrocarbonée, comme un ammonium quaternaire, formant une paire de charge nulle et se partageant entre phases suivant le caractère hydrophobe de l’ensemble. Les résultats dépendent de la nature du contre-ion qui, convenablement choisi, permet des séparations impossibles autrement.La chromatographie par adsorption a été perfectionnée en modifiant chimiquement la phase fixe pour lui donner des propriétés d’échanges très sélectives. Dans la chromatographie par échange de ligands , la silice est combinée à des groupes organiques complexables par un métal de transition, auquel peuvent se lier d’autre part les constituants de la phase mobile. C’est de cette façon qu’on a pu résoudre les mélanges d’énantiomères. La phase fixe comporte des groupes chiraux complexés par du cuivre et les interactions sont différentes avec l’isomère droit et le gauche du même produit (cf. STÉRÉOCHIMIE - Stéréochimie organique).Un principe voisin a présidé à l’élaboration de la chromatographie d’affinité qui permet l’isolement de substances biologiques très spécifiques. Le support solide qui est par exemple un polysaccharide analogue à ceux qu’on emploie en chromatographie sur gel, est chimiquement lié à un ligand biologique présentant une affinité spécifique pour l’un des constituants du mélange tel qu’un sucre, une protéine.Chromatographie sur résines échangeuses d’ionsOn peut également faire appel, en chromatographie, à des phénomènes d’échanges chimiques, pourvu qu’ils soient; comme les échanges physiques précités, réversibles, et qu’ils aient lieu entre deux systèmes physiquement non miscibles. Tel est le cas des réactions d’échange d’ions entre solutions et résines; ils s’agit de polymères insolubles possédant des fonctions acide ou base et façonnés en forme de petites billes de quelques dixièmes de millimètre. Si M+ est un ion métallique dissous, RH la résine, on a la réaction:
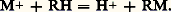 Les ions métalliques sont échangés réversiblement entre les deux phases, suivant les principes régissant les équilibres acides-bases. C’est dire que les paramètres dont on dispose sont la force acide ou basique de la résine échangeuse et les constantes ioniques de la solution, spécialement son pH. Il peut être fixé par addition de substances tampons telles que des phosphates. On peut aussi ajouter des produits complexants, qui modifient la concentration des espèces ioniques libres.Les aminoacides, entre autres, peuvent être séparés par simple régulation du pH de la solution. Considérons-en deux, A et A , dont les points isoélectriques sont respectivement aux pH 5 et 8. On sait qu’en dessous du pH isoélectrique les aminoacides donnent des ions positifs du type de l’ammonium et, au-dessus, des ions carboxyles négatifs. En fixant le pH à 6, l’aminoacide A s’ionise négativement et ne peut s’échanger avec les protons H+ d’une résine acide, alors que pour A , ionisé positivement, cet échange est possible. Les choses sont, en réalité, moins simples que dans ce schéma, mais il n’en reste pas moins qu’on peut ajuster le pH de manière à avoir pour un couple d’aminoacides l’écart de coefficient de partage entre résine et solution le plus favorable à leur séparation. On dispose d’appareils permettant l’analyse automatique de tous les acides aminés d’un échantillon protéinique hydrolysé sur colonnes échangeuses d’ions, dont la composition est donnée directement sur une imprimante. La chromatographie par échange d’ions est évidemment réservée aux substances ionisables, tels les aminoacides et elle est, à ce titre, un procédé de choix en chimie minérale, où elle sert à la séparation de lanthanides ou de produits de fission.Chromatographie d’exclusionQuand une solution est en présence d’un milieu poreux, ses molécules diffusent dans ce milieu, entrant dans les pores et en sortant au hasard. Si les dimensions des molécules dissoutes sont supérieures à celles des pores, elles ne peuvent y pénétrer et en sont exclues. C’est le principe d’une méthode chromatographique qu’on appelle, dans sa version la plus simple, filtration sur gel. La colonne est remplie d’un gel microporeux minéral ou organique: silice, polystyrène réticulé, dextranne réticulé. En fitrant une solution de protéines et de sels minéraux, seules passent les protéines tandis que les sels sont retenus dans la colonne jusqu’à ce qu’elle soit saturée. La séparation est quantitative et très employée pour dessaler des solutions biologiques.La chromatographie par perméation de gel est l’application qualitative. Des molécules de tailles différentes font dans le gel réticulé des parcours de longueur différents, en sortent successivement, et peuvent donc être séparées par ce moyen. Les pores ont des dimensions de l’ordre de celles des molécules moyennes ou grosses. Le procédé s’applique particulièrement bien à l’analyse des macromolécules. L’établissement d’une courbe de répartition des masses d’un polymère, qui auparavant était très laborieuse, se fait en moins d’une heure comme n’importe quelle chromatographie. On dispose pour cela de gels à porosité connue et peu dispersée autour d’une valeur moyenne. L’analyse des copolymères, la recherche des plastifiants ont, grâce à cette technique, énormément progressé.Chromatographie sur papierLe support est ici le papier, comme dans l’expérience de Tswett, mais la véritable phase stationnaire est l’eau d’hydratation des fibres de cellulose et, quand le solvant s’écoule le long de la feuille de papier, c’est un partage qui se produit, entre eau et solvant.La substance à analyser est déposée en une petite tache à une extrémité d’une bande de papier, tache qui se déplace en se séparant en ses composants sous l’effet d’un solvant migrant par capillarité. Les substances colorées sont directement visibles, les autres le sont après pulvérisation sur le papier d’un révélateur. On ne fait pas, en général, d’isolement par élution comme avec une colonne. Les constituants séparés en taches sont caractérisés, puis dosés, par des techniques microchimiques, sur les taches elles-mêmes. On analyse sans peine des échantillons d’un microgramme, mais, avec des traceurs radioactifs, on a pu descendre jusqu’au millionième de microgramme.La séparation est améliorée quand on utilise la chromatographie à deux dimensions. On fait agir successivement sur le papier deux solvants différents dans deux directions perpendiculaires. La tache de mélange déposée en un angle est résolue en plusieurs taches alignées parallèlement à un côté de la feuille par le premier solvant. Le second fait migrer ces taches suivant des lignes parallèles à l’autre côté en les résolvant, si elles sont impures, en leurs composants (fig. 2).Chromatographie sur couches mincesCette technique ressemble à la chromatographie sur papier, la phase fixe étant étalée en une couche mince plane sur une lame de verre. Elle a l’avantage sur la précédente de pouvoir utiliser des phases fixes variées, et notamment des adsorbants, en outre son domaine d’emploi est intermédiaire, en ce qui concerne les quantités, entre la chromatographie en colonne et la chromatographie sur papier. Elle est particulièrement indiquée comme technique d’essai, ses résultats s’extrapolant immédiatement à la technique en colonne puisqu’on utilise les mêmes phases fixes et les mêmes solvants.La préparation des plaques se fait en y étalant une bouillie de l’adsorbant, éventuellement additionné d’un agglomérant. Après séchage, elles sont, comme les feuilles de papier, plongées dans le solvant qui grimpe par capillarité.3. Chromatographie en phase gazeuseLa chromatographie en phase gazeuse utilise, comme l’indique son nom, une phase mobile gazeuse. Les échanges ont lieu entre ce gaz et un solide immobile qui n’a, le plus souvent, d’autre rôle que de servir de support à un liquide qui l’imprègne et constitue la véritable phase fixe. Les constituants du mélange à séparer se partagent entre le gaz vecteur et le liquide dans lequel ils se dissolvent au niveau de chaque grain et l’on a donc, dans ce cas, une résolution basée sur l’équilibre de partage liquide-gaz. Plus rarement, on fait appel à des équilibres d’adsorption gaz-solide: la phase fixe est alors un solide poreux tel que l’alumine ou le carbone activé. Cette dernière technique est la mieux adaptée, notamment, à l’analyse de composés gazeux ou très volatils.Il faut éviter d’employer comme phase mobile un gaz chimiquement actif, vis-à-vis de la phase fixe ou des vapeurs qu’il entraîne; le choix du gaz vecteur est donc très restreint. Pratiquement on se sert d’hélium, d’hydrogène, d’azote ou d’argon. Le mélange à analyser est introduit dans le circuit gazeux par brusque vaporisation dans une chambre d’injection; le tampon de vapeur formé circule dans la colonne au contact de la phase fixe. Comme dans tous les autres procédés, il se fixe sur ses premières sections; puis le gaz vecteur l’entraîne progressivement sur les suivantes, en même temps que la séparation commence à se produire.Un chromatographe en phase vapeur se compose schématiquement d’une colonne dont la température est soigneusement contrôlée, car les propriétés physiques des gaz varient rapidement avec la température, précédée d’un dispositif d’injection des échantillons et suivie d’un détecteur permettant de repérer la sortie des fractions (fig. 3).Dans les procédés d’analyse en isotherme, la température de la colonne est fixe : elle ne doit pas être trop inférieure à la température d’ébullition du constituant le moins volatil du mélange. Si l’on est en présence de composants de volatilités très différentes, il est très avantageux de faire croître la température de colonne au cours de l’analyse suivant un programme préétabli. Les liquides injectés doivent être vaporisés dans la chambre d’injection et rester gazeux dans le détecteur, dont les températures sont stabilisées en conséquence. L’appareil, simple dans son principe, nécessite donc des systèmes de régulation assez complexes.Choix de la phase stationnaireContrairement à ce qui se passe pour la chromatographie en phase liquide, c’est ici le choix de la phase fixe qui conditionne le succès ou l’échec de l’analyse. Le nombre de phases mobiles utilisables est faible; en outre, la composition du gaz vecteur ne change pas pendant la durée d’une analyse, alors qu’en phase liquide on emploie souvent plusieurs solvants.Les phases fixes adsorbantes sont relativement peu nombreuses; leur choix sera fait principalement d’après leur porosité. On dispose par exemple, avec les tamis moléculaires (silicates poreux), de plusieurs qualités dont les pores ont des dimensions du même ordre que celles des molécules et permettent un tri suivant ces dimensions.En chromatographie de partage gaz-liquide, on peut songer à utiliser n’importe quelle phase stationnaire pourvu que soient respectées les conditions évidentes de stabilités physique (non-volatilité à la température de l’expérience) et chimique nécessaires. Aussi, parmi les innombrables produits essayés, plusieurs dizaines sont d’usage courant et l’analyste doit posséder une bonne expérience pour choisir le plus adapté à son problème. Il est également guidé par quelques principes généraux concernant les interactions entre phases mobile et stationnaire.La première notion qui s’impose est celle de la polarité de la phase fixe. Le type d’interaction le plus simple entre vapeur mobile et liquide immobilisé est une dissolution de l’une dans l’autre: la pression d’équilibre est en relation simple avec la tension de vapeur pour chaque constituant du mélange; la chromatographie est alors absolument comparable à une distillation où l’on disposerait d’une colonne de quelques milliers de plateaux théoriques. Tel est le cas pour une phase fixe comme le squalane (hydrocarbure saturé) ou la graisse de silicone et, d’une façon générale, les substances non polaires. Ce sont elles qu’on choisira pour résoudre des mélanges de substances chimiquement voisines et de volatilités différentes, comme les homologues d’une même série organique.Si la phase fixe est polaire, les interactions avec les constituants de la phase mobile sont les mêmes si cette dernière ne contient que des produits non polaires (par exemple un mélange d’hydrocarbures) mais elles sont tout autres s’il y a des produits polaires (par exemple des alcools). Il se forme alors, entre soluté et solvant fixe, des associations par liaisons hydrogènes, retenant fortement les solutés. Ils ne sont élués que bien après des composés non polaires de constantes physiques comparables. Ainsi le benzène et l’éthanol, dont la séparation par distillation est difficile, sont très inégalement retenus par une colonne de polyéthylène glycol: le benzène, entraîné par le gaz vecteur, sort de la colonne bien avant l’alcool éthylique injecté en mélange en même temps que lui. Les spécialistes classent ainsi les différentes phases fixes en fonction de leurs «indices de rétention» pour un échantillonnage de produits témoins. Les plus polaires, comme les esters, alcools ou polyesters lourds, sont adaptés à la séparation de mélanges de produits de polarités différentes.D’autres interactions plus fines peuvent être exploitées: par exemple, les interactions sélectives entre des hydrocarbures benzéniques et des phases fixes contenant elles-mêmes des noyaux aromatiques.On va même jusqu’à la formation de liaisons chimiques faibles entre les deux phases : les savons métalliques sont intéressants pour la séparation des amines.L’appareillageDans la technique la plus classique, les colonnes sont faites de tube métallique de quelques millimètres de diamètre pour une longueur de un à plusieurs mètres, enroulé en spires, afin de limiter l’encombrement. La phase fixe est déposée sur un support granulaire poreux dont elle représente de 10 à 20 p. 100 en poids. La capacité d’une telle colonne, c’est-à-dire la quantité de substance qu’elle peut traiter, avec un bon facteur de séparation, est de l’ordre de quelques dizaines de microlitres au maximum.De plus en plus, ces colonnes sont remplacées par des colonnes capillaires. Longues de 25 à 50 m, allant même jusqu’à 150 m pour un diamètre intérieur de 0,2 à 0,3 mm, elles peuvent ne pas contenir de garnissage poreux. La phase fixe en est la paroi recouverte d’un film de liquide. Avec des tubes un peu plus larges (1 mm), le liquide est déposé sur des grains très fins, eux-mêmes fixés sur les parois. L’efficacité de ces colonnes est de plusieurs dizaines de milliers de plateaux théoriques. Elles travaillent rapidement, séparant un ou deux constituants à la minute; de plus, elles permettent la détection d’une centaine de composants dans un arôme naturel. Mais la quantité de liquide qu’on peut injecter est très faible (une fraction de microlitre) et la dilution, à la sortie, très grande. Cela montre bien la nécessité de disposer de détecteurs très sensibles.Une des raisons du très grand succès de la chromatographie en phase gazeuse est qu’il existe de nombreux procédés de détection des variations de composition d’un effluent gazeux, sensibles et se prêtant bien à une traduction sous forme de signaux électriques. Cela permet même de mettre cette technique d’analyse au service d’unités de fabrication chimique ou de raffinerie. Il suffit de faire des chromatographies à intervalles de temps fixes et de comparer les résultats aux données d’un programme. Le contrôle et le pilotage de l’unité peuvent être entièrement automatisés.Tous les détecteurs ont pour principe la mesure continue d’une propriété physique du gaz sortant de la colonne, dont les variations accompagnent les variations de constitution. Les catharomètres, qui mesurent la conductibilité thermique du gaz, sont les plus employés: le gaz vecteur passe, avant d’entrer dans la colonne, sur un filament chauffé dont la résistance est comparée à celle d’un filament identique balayé par l’effluent. La présence d’une vapeur organique dans l’effluent réduit le refroidissement et les signaux électriques sont traduits en un enregistrement (fig. 4).On décèle les constituants du mélange; de plus on peut également aussi mesurer leurs concentrations relatives à partir de l’aire des «pics» du chromatogramme, qui, en première approximation, en dépend linéairement.On emploie aussi très couramment des détecteurs à ionisation de flamme, basés sur la formation d’ions dans une flamme où passent des composés hydrocarbonés, les détecteurs à ionisation d’argon et à capture d’électrons. Des concentrations aussi basses qu’une partie de substance organique pour 1010 de gaz vecteur sont décelables.Le couplage de la chromatographie avec la spectrographie de masse est fait par de nombreux constructeurs et permet, outre la détection des fractions, l’identification des produits par leur spectre de masse ou simplement par son pic le plus lourd.La chromatographie en phase gazeuse ayant une efficacité très grande comme moyen de séparation, on a tenté de l’étendre à la préparation de quantités appréciables de substances. On est limité dans l’accroissement de dimensions des colonnes qui se fait aux dépens de la qualité, mais on dispose tout de même d’appareils capables de fournir, généralement par répétition de cycles identiques, quelques grammes de produits purs.La chromatographie préparative en phase gazeuse est devenue courante au laboratoire sur des colonnes de l’ordre du centimètre de diamètre. Les colonnes industrielles perdent en efficacité; toutefois, avec quelque mille plateaux théoriques au mètre, elles sont encore très supérieures aux colonnes de distillation et moins coûteuses en énergie. La première unité, pour la préparation de parfums, a démarré aux États-Unis avec une productivité de 100 tonnes par an.4. Chromatographie en phase supercritiqueLa chromatographie en phase supercritique (C.P.S.) met en œuvre, comme phase mobile, un fluide porté au-delà de l’état critique par un contrôle adéquat de la température et de la pression.On peut représenter les trois états classiques de la matière – solide S, liquide L, vapeur V – dans un diagramme pression-température (fig. 5). Lorsqu’on passe d’un domaine à l’autre, généralement par une variation de température (en chauffant ou en refroidissant), il y a une discontinuité de la masse volumique, lors de la sublimation (SV), de la fusion (SL) ou de l’ébullition (LV). Au point triple T coexistent les trois phases solide-liquide-vapeur.Lors de la fusion, il n’est pas possible de passer d’un état à l’autre sans traverser un segment; mais on peut passer de l’état liquide à l’état gazeux continûment, sans franchir le segment L-V, à la condition de contourner le point C où se termine la courbe d’équilibre liquide-vapeur et qui est appelé point critique. À ce dernier correspond une température critique c, une pression critique Pc et une masse volumique critique 福c. Au-delà de C, on a un état intermédiaire qu’on appelle état critique ou, si l’on s’éloigne notablement de C, état supercritique.Il est intéressant de considérer le diagramme de phase (P, 福) du dioxyde de carbone. Plusieurs isothermes ont été tracés et la zone d’utilisation habituelle de la C.P.S. a été grisée (fig. 6). Lorsque la température est inférieure à c, on observe la portion du domaine d’équilibre liquide-vapeur où les masses volumiques de ces deux phases sont différentes. Au fur et à mesure que l’on se rapproche de l’isotherme critique, cette différence diminue, pour s’annuler lorsque c est atteinte. Sur l’isotherme critique, le palier se réduit au point critique. Le passage de l’état liquide à l’état gazeux se fait à une pression, appelée pression de vapeur saturante, qui ne dépend que de la température.Les propriétés des fluides supercritiques sont très particulières. Ainsi, à température constante, la masse volumique augmente avec la pression; on choisit des conditions de température et de pression correspondant à des valeurs élevées, comprises entre 0,4 et 1 g. cm-3: la masse volumique d’un fluide supercritique est donc beaucoup plus proche de celle d’un liquide que de celle d’un gaz, ce qui lui permet d’avoir ainsi un pouvoir solvant.En revanche, la viscosité 兀 d’un fluide supercritique (qui augmente avec la pression et diminue lorsque la température monte) est comprise entre 10-4 et 10-3 Pa . s: supérieure à celle des gaz, elle est de dix à cent fois inférieure à celle des liquides. Il en résulte que la diffusion d’un soluté sera beaucoup plus rapide dans un fluide supercritique que dans un liquide. On a pu dire qu’un fluide supercritique était analogue au «métro aux heures de pointe»: la densité des usagers y est élevée et, pourtant, le milieu reste fluide et les déplacements rapides.Ainsi, l’intérêt d’utiliser un fluide supercritique en chromatographie réside dans les caractéristiques suivantes:– possibilité d’y dissoudre des solutés variés;– séparation de composés thermosensibles dans la mesure où la température, à l’état supercritique, reste modérée (par exemple 40-90 0C dans le cas du C2);– meilleure efficacité du système chromatographique en raison de la faible viscosité du fluide supercritique: le nombre de plateaux théoriques, par unité de temps, est très supérieur à celui qui est obtenu avec les liquides; les séparations pourront être très rapides dans le cas des colonnes remplies (quelques minutes);– possibilité de mettre en œuvre, comme en chromatographie en phase gazeuse, des colonnes capillaires de grande longueur (plusieurs dizaines de mètres): on dispose alors de centaines de milliers de plateaux théoriques qui autorisent la séparation de mélanges très complexes: oligomères, esters d’acide gras, additifs de polymères, etc.;– enfin, les méthodes de détection de la chromatographie en phase supercritique peuvent être associées soit à celles de la chromatographie en phase liquide, soit à celles de la chromatographie en phase gazeuse; par exemple, le C2 est transparent dans l’ultraviolet, d’où une détection possible par absorptiométrie U.V. (comme en liquide); il a aussi de larges zones de transparence dans l’infrarouge, d’où la possibilité de coupler la C.P.S. avec la spectrométrie infrarouge à transformée de Fourier (I.R.T.F.); on peut alors tracer le spectre infrarouge des solutés au fur et à mesure qu’ils émergent de la colonne chromatographique, ce qui offre de remarquables possibilités d’identification, dans le cas de solutés inconnus, au moyen de bibliothèques de spectres.Le dioxyde de carbone C2 permet une détection par ionisation de flamme (comme en chromatographie en phase gazeuse), qui est à la fois universelle, sensible et peu dépendante de la nature des solutés, ce qui facilite l’analyse quantitative. On a également couplé la C.P.S. avec la fluorimétrie, la spectrométrie de masse (méthode d’identification absolue des solutés par leur spectre de masse) et la diffusion de la lumière.Le dioxyde de carbone est, de loin, le fluide le plus utilisé en C.P.S. en raison de nombreuses propriétés intéressantes: sa manipulation est facile, compte tenu de sa température et de sa pression critiques (31,0 0C, 73,8 bars); sa température critique est compatible avec la stabilité des solutés et des silices greffées; il a un bon pouvoir solvant et est non toxique, ininflammable, non corrosif, inodore et peu onéreux (environ 9 F le litre de C2 liquide dans la qualité technique, 75 F le litre dans la qualité N45-99,995 p. 100 de pureté, prix H.T. en 1988); il est disponible, si cela est nécessaire, dans une très grande pureté (mais son prix augmente alors d’un facteur 8 pour la qualité N45); enfin, la récupération des solutés, en chromatographie préparative, est aisée (par détente ou par réchauffement).Toutefois, d’autres fluides sont utilisés: des alcanes comme le propane, le butane ou le pentane (mais des risques d’inflammation et d’explosion existent, de sorte qu’on n’utilise que les colonnes capillaires qui limitent les volumes de solvant), des solvants fluorés – hexafluorure de soufre, fréon – et le xénon, très onéreux, mais qui est totalement transparent quelle que soit la longueur d’onde (obtention du spectre complet en infrarouge).Propriétés chromatographiquesAvec le C2 pur, on peut faire varier la rétention en intervenant sur la masse volumique par la température et la pression. Cette possibilité d’agir sur les interactions par l’état du fluide est très riche: d’une manière générale, une augmentation de la masse volumique diminue la rétention.Par ailleurs, le C2 est miscible en toute proportion avec les solvants organiques qui, ajoutés en faibles quantités au C2 sont des modificateurs polaires. Ils diminuent la rétention et, surtout, entraînent des variations de sélectivité qui offrent de multiples possibilités.L’appareillage en chromatographie en phase supercritiqueSeules quelques indications sont données ici. Deux points sont à souligner:– la mise en œuvre des fluides supercritiques nécessite un contrôle strict de la pression plutôt qu’un contrôle du débit;– l’état supercritique, ou tout au moins l’état liquide, doit être maintenu jusque dans le système de détection lorsqu’on utilise un spectrophotomètre et celui-ci doit être conçu pour résister à des pressions de l’ordre de 350 bars dans le cas du dioxyde de carbone; en effet, la détection sous pression atmosphérique est impossible, car la solubilité des solutés dans les gaz est très faible, d’où une précipitation de ceux-ci dans le système de détection, ce qui provoque un signal erratique; en revanche, on détend le fluide supercritique en ionisation de flamme ou en spectrométrie de masse.Le schéma général d’un chromatographe, en phase supercritique, est représenté à la figure 7.Le dioxyde de carbone est contenu dans un réservoir muni d’un tube plongeur. La pression d’équilibre, à la température ambiante, est d’environ 55 bars. Pour que le pompage soit efficace, le fluide doit être à l’état liquide: pour ce faire, on refroidit le corps de pompe à 0 0C par une circulation d’éthanol. L’addition d’un modificateur polaire, comme le méthanol au C2, se fait au moyen d’une pompe à piston capable de délivrer de très faibles débits (jusqu’à 5 猪l. min-1). Les systèmes d’injection à boucle sont analogues à ceux de la chromatographie en phase liquide. Le contrôle de la température de la colonne, de l’injecteur et du détecteur est assuré par un bain thermorégulé ou par un four.Dans le cas des colonnes remplies, des vannes spéciales régulent la pression indépendamment du débit du fluide. Celles-ci sont placées après le détecteur. Avec les colonnes capillaires, la régulation se fait au moyen d’un restricteur qui exige une technologie particulière. De nombreux chromatographes en phase supercritique sont commercialisés.Exemples d’applicationsUn domaine important de la C.P.S. consiste en la séparation des oligomères des polymères, comme ceux de polystyrènes. Par une variation régulière de la pression au cours de la séparation (ce qu’on appelle une programmation de pression), on parvient à séparer les différents oligomères de l’échantillon, c’est-à-dire que des molécules diffèrent entre elles seulement par un motif styrène.La figure 8 montre une séparation particulièrement difficile, celle des additifs utilisés pour stabiliser les polymères. La détection est effectuée par ionisation de flamme mais on peut aussi, chaque fois qu’un pic émerge de la colonne, le recueillir sur une pastille de bromure de potassium, introduire celle-ci dans un microscope associé à un spectromètre à transformée de Fourier et obtenir ainsi le spectre infrarouge du composé. Cela permet à la fois d’identifier avec sûreté les additifs mais aussi de suivre leur évolution au fur et à mesure du vieillissement du polymère dans des conditions données.Parmi les autres domaines d’application explorés, citons l’analyse des produits pétroliers (séparation en familles: alcanes, oléfines, aromatiques, composés polaires), des acides gras, des alcaloïdes morphiniques, des terpénoïdes, de composés chiraux, etc. Une possibilité attractive de la C.P.S. est qu’elle se prête bien, au moins théoriquement, aux séparations à l’échelle préparative: il suffit de détendre et/ou de réchauffer le fluide supercritique pour obtenir son élimination d’une manière efficace et rapide. Toutefois, la sédimentation des «brouillards» obtenus par détente pose de délicats problèmes technologiques.La chromatographie en phase supercritique témoigne des progrès obtenus depuis la découverte de l’état supercritique par le baron Charles Cagniard de La Tour (1777-1859) et les expériences mémorables de Hanay et Hogarth montrant que les chlorures de fer et de cobalt sont solubles dans l’éthanol supercritique. Cette méthode a sa place dans les laboratoires d’analyse pour 25 p. 100 des substances connues, où elle complète les chromatographies plus classiques en phase liquide ou gazeuse.
Les ions métalliques sont échangés réversiblement entre les deux phases, suivant les principes régissant les équilibres acides-bases. C’est dire que les paramètres dont on dispose sont la force acide ou basique de la résine échangeuse et les constantes ioniques de la solution, spécialement son pH. Il peut être fixé par addition de substances tampons telles que des phosphates. On peut aussi ajouter des produits complexants, qui modifient la concentration des espèces ioniques libres.Les aminoacides, entre autres, peuvent être séparés par simple régulation du pH de la solution. Considérons-en deux, A et A , dont les points isoélectriques sont respectivement aux pH 5 et 8. On sait qu’en dessous du pH isoélectrique les aminoacides donnent des ions positifs du type de l’ammonium et, au-dessus, des ions carboxyles négatifs. En fixant le pH à 6, l’aminoacide A s’ionise négativement et ne peut s’échanger avec les protons H+ d’une résine acide, alors que pour A , ionisé positivement, cet échange est possible. Les choses sont, en réalité, moins simples que dans ce schéma, mais il n’en reste pas moins qu’on peut ajuster le pH de manière à avoir pour un couple d’aminoacides l’écart de coefficient de partage entre résine et solution le plus favorable à leur séparation. On dispose d’appareils permettant l’analyse automatique de tous les acides aminés d’un échantillon protéinique hydrolysé sur colonnes échangeuses d’ions, dont la composition est donnée directement sur une imprimante. La chromatographie par échange d’ions est évidemment réservée aux substances ionisables, tels les aminoacides et elle est, à ce titre, un procédé de choix en chimie minérale, où elle sert à la séparation de lanthanides ou de produits de fission.Chromatographie d’exclusionQuand une solution est en présence d’un milieu poreux, ses molécules diffusent dans ce milieu, entrant dans les pores et en sortant au hasard. Si les dimensions des molécules dissoutes sont supérieures à celles des pores, elles ne peuvent y pénétrer et en sont exclues. C’est le principe d’une méthode chromatographique qu’on appelle, dans sa version la plus simple, filtration sur gel. La colonne est remplie d’un gel microporeux minéral ou organique: silice, polystyrène réticulé, dextranne réticulé. En fitrant une solution de protéines et de sels minéraux, seules passent les protéines tandis que les sels sont retenus dans la colonne jusqu’à ce qu’elle soit saturée. La séparation est quantitative et très employée pour dessaler des solutions biologiques.La chromatographie par perméation de gel est l’application qualitative. Des molécules de tailles différentes font dans le gel réticulé des parcours de longueur différents, en sortent successivement, et peuvent donc être séparées par ce moyen. Les pores ont des dimensions de l’ordre de celles des molécules moyennes ou grosses. Le procédé s’applique particulièrement bien à l’analyse des macromolécules. L’établissement d’une courbe de répartition des masses d’un polymère, qui auparavant était très laborieuse, se fait en moins d’une heure comme n’importe quelle chromatographie. On dispose pour cela de gels à porosité connue et peu dispersée autour d’une valeur moyenne. L’analyse des copolymères, la recherche des plastifiants ont, grâce à cette technique, énormément progressé.Chromatographie sur papierLe support est ici le papier, comme dans l’expérience de Tswett, mais la véritable phase stationnaire est l’eau d’hydratation des fibres de cellulose et, quand le solvant s’écoule le long de la feuille de papier, c’est un partage qui se produit, entre eau et solvant.La substance à analyser est déposée en une petite tache à une extrémité d’une bande de papier, tache qui se déplace en se séparant en ses composants sous l’effet d’un solvant migrant par capillarité. Les substances colorées sont directement visibles, les autres le sont après pulvérisation sur le papier d’un révélateur. On ne fait pas, en général, d’isolement par élution comme avec une colonne. Les constituants séparés en taches sont caractérisés, puis dosés, par des techniques microchimiques, sur les taches elles-mêmes. On analyse sans peine des échantillons d’un microgramme, mais, avec des traceurs radioactifs, on a pu descendre jusqu’au millionième de microgramme.La séparation est améliorée quand on utilise la chromatographie à deux dimensions. On fait agir successivement sur le papier deux solvants différents dans deux directions perpendiculaires. La tache de mélange déposée en un angle est résolue en plusieurs taches alignées parallèlement à un côté de la feuille par le premier solvant. Le second fait migrer ces taches suivant des lignes parallèles à l’autre côté en les résolvant, si elles sont impures, en leurs composants (fig. 2).Chromatographie sur couches mincesCette technique ressemble à la chromatographie sur papier, la phase fixe étant étalée en une couche mince plane sur une lame de verre. Elle a l’avantage sur la précédente de pouvoir utiliser des phases fixes variées, et notamment des adsorbants, en outre son domaine d’emploi est intermédiaire, en ce qui concerne les quantités, entre la chromatographie en colonne et la chromatographie sur papier. Elle est particulièrement indiquée comme technique d’essai, ses résultats s’extrapolant immédiatement à la technique en colonne puisqu’on utilise les mêmes phases fixes et les mêmes solvants.La préparation des plaques se fait en y étalant une bouillie de l’adsorbant, éventuellement additionné d’un agglomérant. Après séchage, elles sont, comme les feuilles de papier, plongées dans le solvant qui grimpe par capillarité.3. Chromatographie en phase gazeuseLa chromatographie en phase gazeuse utilise, comme l’indique son nom, une phase mobile gazeuse. Les échanges ont lieu entre ce gaz et un solide immobile qui n’a, le plus souvent, d’autre rôle que de servir de support à un liquide qui l’imprègne et constitue la véritable phase fixe. Les constituants du mélange à séparer se partagent entre le gaz vecteur et le liquide dans lequel ils se dissolvent au niveau de chaque grain et l’on a donc, dans ce cas, une résolution basée sur l’équilibre de partage liquide-gaz. Plus rarement, on fait appel à des équilibres d’adsorption gaz-solide: la phase fixe est alors un solide poreux tel que l’alumine ou le carbone activé. Cette dernière technique est la mieux adaptée, notamment, à l’analyse de composés gazeux ou très volatils.Il faut éviter d’employer comme phase mobile un gaz chimiquement actif, vis-à-vis de la phase fixe ou des vapeurs qu’il entraîne; le choix du gaz vecteur est donc très restreint. Pratiquement on se sert d’hélium, d’hydrogène, d’azote ou d’argon. Le mélange à analyser est introduit dans le circuit gazeux par brusque vaporisation dans une chambre d’injection; le tampon de vapeur formé circule dans la colonne au contact de la phase fixe. Comme dans tous les autres procédés, il se fixe sur ses premières sections; puis le gaz vecteur l’entraîne progressivement sur les suivantes, en même temps que la séparation commence à se produire.Un chromatographe en phase vapeur se compose schématiquement d’une colonne dont la température est soigneusement contrôlée, car les propriétés physiques des gaz varient rapidement avec la température, précédée d’un dispositif d’injection des échantillons et suivie d’un détecteur permettant de repérer la sortie des fractions (fig. 3).Dans les procédés d’analyse en isotherme, la température de la colonne est fixe : elle ne doit pas être trop inférieure à la température d’ébullition du constituant le moins volatil du mélange. Si l’on est en présence de composants de volatilités très différentes, il est très avantageux de faire croître la température de colonne au cours de l’analyse suivant un programme préétabli. Les liquides injectés doivent être vaporisés dans la chambre d’injection et rester gazeux dans le détecteur, dont les températures sont stabilisées en conséquence. L’appareil, simple dans son principe, nécessite donc des systèmes de régulation assez complexes.Choix de la phase stationnaireContrairement à ce qui se passe pour la chromatographie en phase liquide, c’est ici le choix de la phase fixe qui conditionne le succès ou l’échec de l’analyse. Le nombre de phases mobiles utilisables est faible; en outre, la composition du gaz vecteur ne change pas pendant la durée d’une analyse, alors qu’en phase liquide on emploie souvent plusieurs solvants.Les phases fixes adsorbantes sont relativement peu nombreuses; leur choix sera fait principalement d’après leur porosité. On dispose par exemple, avec les tamis moléculaires (silicates poreux), de plusieurs qualités dont les pores ont des dimensions du même ordre que celles des molécules et permettent un tri suivant ces dimensions.En chromatographie de partage gaz-liquide, on peut songer à utiliser n’importe quelle phase stationnaire pourvu que soient respectées les conditions évidentes de stabilités physique (non-volatilité à la température de l’expérience) et chimique nécessaires. Aussi, parmi les innombrables produits essayés, plusieurs dizaines sont d’usage courant et l’analyste doit posséder une bonne expérience pour choisir le plus adapté à son problème. Il est également guidé par quelques principes généraux concernant les interactions entre phases mobile et stationnaire.La première notion qui s’impose est celle de la polarité de la phase fixe. Le type d’interaction le plus simple entre vapeur mobile et liquide immobilisé est une dissolution de l’une dans l’autre: la pression d’équilibre est en relation simple avec la tension de vapeur pour chaque constituant du mélange; la chromatographie est alors absolument comparable à une distillation où l’on disposerait d’une colonne de quelques milliers de plateaux théoriques. Tel est le cas pour une phase fixe comme le squalane (hydrocarbure saturé) ou la graisse de silicone et, d’une façon générale, les substances non polaires. Ce sont elles qu’on choisira pour résoudre des mélanges de substances chimiquement voisines et de volatilités différentes, comme les homologues d’une même série organique.Si la phase fixe est polaire, les interactions avec les constituants de la phase mobile sont les mêmes si cette dernière ne contient que des produits non polaires (par exemple un mélange d’hydrocarbures) mais elles sont tout autres s’il y a des produits polaires (par exemple des alcools). Il se forme alors, entre soluté et solvant fixe, des associations par liaisons hydrogènes, retenant fortement les solutés. Ils ne sont élués que bien après des composés non polaires de constantes physiques comparables. Ainsi le benzène et l’éthanol, dont la séparation par distillation est difficile, sont très inégalement retenus par une colonne de polyéthylène glycol: le benzène, entraîné par le gaz vecteur, sort de la colonne bien avant l’alcool éthylique injecté en mélange en même temps que lui. Les spécialistes classent ainsi les différentes phases fixes en fonction de leurs «indices de rétention» pour un échantillonnage de produits témoins. Les plus polaires, comme les esters, alcools ou polyesters lourds, sont adaptés à la séparation de mélanges de produits de polarités différentes.D’autres interactions plus fines peuvent être exploitées: par exemple, les interactions sélectives entre des hydrocarbures benzéniques et des phases fixes contenant elles-mêmes des noyaux aromatiques.On va même jusqu’à la formation de liaisons chimiques faibles entre les deux phases : les savons métalliques sont intéressants pour la séparation des amines.L’appareillageDans la technique la plus classique, les colonnes sont faites de tube métallique de quelques millimètres de diamètre pour une longueur de un à plusieurs mètres, enroulé en spires, afin de limiter l’encombrement. La phase fixe est déposée sur un support granulaire poreux dont elle représente de 10 à 20 p. 100 en poids. La capacité d’une telle colonne, c’est-à-dire la quantité de substance qu’elle peut traiter, avec un bon facteur de séparation, est de l’ordre de quelques dizaines de microlitres au maximum.De plus en plus, ces colonnes sont remplacées par des colonnes capillaires. Longues de 25 à 50 m, allant même jusqu’à 150 m pour un diamètre intérieur de 0,2 à 0,3 mm, elles peuvent ne pas contenir de garnissage poreux. La phase fixe en est la paroi recouverte d’un film de liquide. Avec des tubes un peu plus larges (1 mm), le liquide est déposé sur des grains très fins, eux-mêmes fixés sur les parois. L’efficacité de ces colonnes est de plusieurs dizaines de milliers de plateaux théoriques. Elles travaillent rapidement, séparant un ou deux constituants à la minute; de plus, elles permettent la détection d’une centaine de composants dans un arôme naturel. Mais la quantité de liquide qu’on peut injecter est très faible (une fraction de microlitre) et la dilution, à la sortie, très grande. Cela montre bien la nécessité de disposer de détecteurs très sensibles.Une des raisons du très grand succès de la chromatographie en phase gazeuse est qu’il existe de nombreux procédés de détection des variations de composition d’un effluent gazeux, sensibles et se prêtant bien à une traduction sous forme de signaux électriques. Cela permet même de mettre cette technique d’analyse au service d’unités de fabrication chimique ou de raffinerie. Il suffit de faire des chromatographies à intervalles de temps fixes et de comparer les résultats aux données d’un programme. Le contrôle et le pilotage de l’unité peuvent être entièrement automatisés.Tous les détecteurs ont pour principe la mesure continue d’une propriété physique du gaz sortant de la colonne, dont les variations accompagnent les variations de constitution. Les catharomètres, qui mesurent la conductibilité thermique du gaz, sont les plus employés: le gaz vecteur passe, avant d’entrer dans la colonne, sur un filament chauffé dont la résistance est comparée à celle d’un filament identique balayé par l’effluent. La présence d’une vapeur organique dans l’effluent réduit le refroidissement et les signaux électriques sont traduits en un enregistrement (fig. 4).On décèle les constituants du mélange; de plus on peut également aussi mesurer leurs concentrations relatives à partir de l’aire des «pics» du chromatogramme, qui, en première approximation, en dépend linéairement.On emploie aussi très couramment des détecteurs à ionisation de flamme, basés sur la formation d’ions dans une flamme où passent des composés hydrocarbonés, les détecteurs à ionisation d’argon et à capture d’électrons. Des concentrations aussi basses qu’une partie de substance organique pour 1010 de gaz vecteur sont décelables.Le couplage de la chromatographie avec la spectrographie de masse est fait par de nombreux constructeurs et permet, outre la détection des fractions, l’identification des produits par leur spectre de masse ou simplement par son pic le plus lourd.La chromatographie en phase gazeuse ayant une efficacité très grande comme moyen de séparation, on a tenté de l’étendre à la préparation de quantités appréciables de substances. On est limité dans l’accroissement de dimensions des colonnes qui se fait aux dépens de la qualité, mais on dispose tout de même d’appareils capables de fournir, généralement par répétition de cycles identiques, quelques grammes de produits purs.La chromatographie préparative en phase gazeuse est devenue courante au laboratoire sur des colonnes de l’ordre du centimètre de diamètre. Les colonnes industrielles perdent en efficacité; toutefois, avec quelque mille plateaux théoriques au mètre, elles sont encore très supérieures aux colonnes de distillation et moins coûteuses en énergie. La première unité, pour la préparation de parfums, a démarré aux États-Unis avec une productivité de 100 tonnes par an.4. Chromatographie en phase supercritiqueLa chromatographie en phase supercritique (C.P.S.) met en œuvre, comme phase mobile, un fluide porté au-delà de l’état critique par un contrôle adéquat de la température et de la pression.On peut représenter les trois états classiques de la matière – solide S, liquide L, vapeur V – dans un diagramme pression-température (fig. 5). Lorsqu’on passe d’un domaine à l’autre, généralement par une variation de température (en chauffant ou en refroidissant), il y a une discontinuité de la masse volumique, lors de la sublimation (SV), de la fusion (SL) ou de l’ébullition (LV). Au point triple T coexistent les trois phases solide-liquide-vapeur.Lors de la fusion, il n’est pas possible de passer d’un état à l’autre sans traverser un segment; mais on peut passer de l’état liquide à l’état gazeux continûment, sans franchir le segment L-V, à la condition de contourner le point C où se termine la courbe d’équilibre liquide-vapeur et qui est appelé point critique. À ce dernier correspond une température critique c, une pression critique Pc et une masse volumique critique 福c. Au-delà de C, on a un état intermédiaire qu’on appelle état critique ou, si l’on s’éloigne notablement de C, état supercritique.Il est intéressant de considérer le diagramme de phase (P, 福) du dioxyde de carbone. Plusieurs isothermes ont été tracés et la zone d’utilisation habituelle de la C.P.S. a été grisée (fig. 6). Lorsque la température est inférieure à c, on observe la portion du domaine d’équilibre liquide-vapeur où les masses volumiques de ces deux phases sont différentes. Au fur et à mesure que l’on se rapproche de l’isotherme critique, cette différence diminue, pour s’annuler lorsque c est atteinte. Sur l’isotherme critique, le palier se réduit au point critique. Le passage de l’état liquide à l’état gazeux se fait à une pression, appelée pression de vapeur saturante, qui ne dépend que de la température.Les propriétés des fluides supercritiques sont très particulières. Ainsi, à température constante, la masse volumique augmente avec la pression; on choisit des conditions de température et de pression correspondant à des valeurs élevées, comprises entre 0,4 et 1 g. cm-3: la masse volumique d’un fluide supercritique est donc beaucoup plus proche de celle d’un liquide que de celle d’un gaz, ce qui lui permet d’avoir ainsi un pouvoir solvant.En revanche, la viscosité 兀 d’un fluide supercritique (qui augmente avec la pression et diminue lorsque la température monte) est comprise entre 10-4 et 10-3 Pa . s: supérieure à celle des gaz, elle est de dix à cent fois inférieure à celle des liquides. Il en résulte que la diffusion d’un soluté sera beaucoup plus rapide dans un fluide supercritique que dans un liquide. On a pu dire qu’un fluide supercritique était analogue au «métro aux heures de pointe»: la densité des usagers y est élevée et, pourtant, le milieu reste fluide et les déplacements rapides.Ainsi, l’intérêt d’utiliser un fluide supercritique en chromatographie réside dans les caractéristiques suivantes:– possibilité d’y dissoudre des solutés variés;– séparation de composés thermosensibles dans la mesure où la température, à l’état supercritique, reste modérée (par exemple 40-90 0C dans le cas du C2);– meilleure efficacité du système chromatographique en raison de la faible viscosité du fluide supercritique: le nombre de plateaux théoriques, par unité de temps, est très supérieur à celui qui est obtenu avec les liquides; les séparations pourront être très rapides dans le cas des colonnes remplies (quelques minutes);– possibilité de mettre en œuvre, comme en chromatographie en phase gazeuse, des colonnes capillaires de grande longueur (plusieurs dizaines de mètres): on dispose alors de centaines de milliers de plateaux théoriques qui autorisent la séparation de mélanges très complexes: oligomères, esters d’acide gras, additifs de polymères, etc.;– enfin, les méthodes de détection de la chromatographie en phase supercritique peuvent être associées soit à celles de la chromatographie en phase liquide, soit à celles de la chromatographie en phase gazeuse; par exemple, le C2 est transparent dans l’ultraviolet, d’où une détection possible par absorptiométrie U.V. (comme en liquide); il a aussi de larges zones de transparence dans l’infrarouge, d’où la possibilité de coupler la C.P.S. avec la spectrométrie infrarouge à transformée de Fourier (I.R.T.F.); on peut alors tracer le spectre infrarouge des solutés au fur et à mesure qu’ils émergent de la colonne chromatographique, ce qui offre de remarquables possibilités d’identification, dans le cas de solutés inconnus, au moyen de bibliothèques de spectres.Le dioxyde de carbone C2 permet une détection par ionisation de flamme (comme en chromatographie en phase gazeuse), qui est à la fois universelle, sensible et peu dépendante de la nature des solutés, ce qui facilite l’analyse quantitative. On a également couplé la C.P.S. avec la fluorimétrie, la spectrométrie de masse (méthode d’identification absolue des solutés par leur spectre de masse) et la diffusion de la lumière.Le dioxyde de carbone est, de loin, le fluide le plus utilisé en C.P.S. en raison de nombreuses propriétés intéressantes: sa manipulation est facile, compte tenu de sa température et de sa pression critiques (31,0 0C, 73,8 bars); sa température critique est compatible avec la stabilité des solutés et des silices greffées; il a un bon pouvoir solvant et est non toxique, ininflammable, non corrosif, inodore et peu onéreux (environ 9 F le litre de C2 liquide dans la qualité technique, 75 F le litre dans la qualité N45-99,995 p. 100 de pureté, prix H.T. en 1988); il est disponible, si cela est nécessaire, dans une très grande pureté (mais son prix augmente alors d’un facteur 8 pour la qualité N45); enfin, la récupération des solutés, en chromatographie préparative, est aisée (par détente ou par réchauffement).Toutefois, d’autres fluides sont utilisés: des alcanes comme le propane, le butane ou le pentane (mais des risques d’inflammation et d’explosion existent, de sorte qu’on n’utilise que les colonnes capillaires qui limitent les volumes de solvant), des solvants fluorés – hexafluorure de soufre, fréon – et le xénon, très onéreux, mais qui est totalement transparent quelle que soit la longueur d’onde (obtention du spectre complet en infrarouge).Propriétés chromatographiquesAvec le C2 pur, on peut faire varier la rétention en intervenant sur la masse volumique par la température et la pression. Cette possibilité d’agir sur les interactions par l’état du fluide est très riche: d’une manière générale, une augmentation de la masse volumique diminue la rétention.Par ailleurs, le C2 est miscible en toute proportion avec les solvants organiques qui, ajoutés en faibles quantités au C2 sont des modificateurs polaires. Ils diminuent la rétention et, surtout, entraînent des variations de sélectivité qui offrent de multiples possibilités.L’appareillage en chromatographie en phase supercritiqueSeules quelques indications sont données ici. Deux points sont à souligner:– la mise en œuvre des fluides supercritiques nécessite un contrôle strict de la pression plutôt qu’un contrôle du débit;– l’état supercritique, ou tout au moins l’état liquide, doit être maintenu jusque dans le système de détection lorsqu’on utilise un spectrophotomètre et celui-ci doit être conçu pour résister à des pressions de l’ordre de 350 bars dans le cas du dioxyde de carbone; en effet, la détection sous pression atmosphérique est impossible, car la solubilité des solutés dans les gaz est très faible, d’où une précipitation de ceux-ci dans le système de détection, ce qui provoque un signal erratique; en revanche, on détend le fluide supercritique en ionisation de flamme ou en spectrométrie de masse.Le schéma général d’un chromatographe, en phase supercritique, est représenté à la figure 7.Le dioxyde de carbone est contenu dans un réservoir muni d’un tube plongeur. La pression d’équilibre, à la température ambiante, est d’environ 55 bars. Pour que le pompage soit efficace, le fluide doit être à l’état liquide: pour ce faire, on refroidit le corps de pompe à 0 0C par une circulation d’éthanol. L’addition d’un modificateur polaire, comme le méthanol au C2, se fait au moyen d’une pompe à piston capable de délivrer de très faibles débits (jusqu’à 5 猪l. min-1). Les systèmes d’injection à boucle sont analogues à ceux de la chromatographie en phase liquide. Le contrôle de la température de la colonne, de l’injecteur et du détecteur est assuré par un bain thermorégulé ou par un four.Dans le cas des colonnes remplies, des vannes spéciales régulent la pression indépendamment du débit du fluide. Celles-ci sont placées après le détecteur. Avec les colonnes capillaires, la régulation se fait au moyen d’un restricteur qui exige une technologie particulière. De nombreux chromatographes en phase supercritique sont commercialisés.Exemples d’applicationsUn domaine important de la C.P.S. consiste en la séparation des oligomères des polymères, comme ceux de polystyrènes. Par une variation régulière de la pression au cours de la séparation (ce qu’on appelle une programmation de pression), on parvient à séparer les différents oligomères de l’échantillon, c’est-à-dire que des molécules diffèrent entre elles seulement par un motif styrène.La figure 8 montre une séparation particulièrement difficile, celle des additifs utilisés pour stabiliser les polymères. La détection est effectuée par ionisation de flamme mais on peut aussi, chaque fois qu’un pic émerge de la colonne, le recueillir sur une pastille de bromure de potassium, introduire celle-ci dans un microscope associé à un spectromètre à transformée de Fourier et obtenir ainsi le spectre infrarouge du composé. Cela permet à la fois d’identifier avec sûreté les additifs mais aussi de suivre leur évolution au fur et à mesure du vieillissement du polymère dans des conditions données.Parmi les autres domaines d’application explorés, citons l’analyse des produits pétroliers (séparation en familles: alcanes, oléfines, aromatiques, composés polaires), des acides gras, des alcaloïdes morphiniques, des terpénoïdes, de composés chiraux, etc. Une possibilité attractive de la C.P.S. est qu’elle se prête bien, au moins théoriquement, aux séparations à l’échelle préparative: il suffit de détendre et/ou de réchauffer le fluide supercritique pour obtenir son élimination d’une manière efficace et rapide. Toutefois, la sédimentation des «brouillards» obtenus par détente pose de délicats problèmes technologiques.La chromatographie en phase supercritique témoigne des progrès obtenus depuis la découverte de l’état supercritique par le baron Charles Cagniard de La Tour (1777-1859) et les expériences mémorables de Hanay et Hogarth montrant que les chlorures de fer et de cobalt sont solubles dans l’éthanol supercritique. Cette méthode a sa place dans les laboratoires d’analyse pour 25 p. 100 des substances connues, où elle complète les chromatographies plus classiques en phase liquide ou gazeuse.
chromatographie [ krɔmatɔgrafi ] n. f. ♦ Chim. Méthode d'analyse chimique et de purification des constituants d'un mélange par adsorption sélective des constituants du mélange, ou par partage en présence de phases liquides ou gazeuses (⇒ élution).
● chromatographie nom féminin Technique permettant de séparer les constituants d'un mélange afin de les doser.chromatographien. f. BIOCHIM Procédé de séparation de différentes substances en solution ou en suspension dans un liquide.⇒CHROMATOGRAPHIE, subst. fém.CHIM. ,,Technique qui, faisant appel à l'adsorption sélective par diverses substances pulvérulentes ou par des papiers, permet de séparer les différents constituants d'un mélange complexe, en particulier de substances colorantes`` (HUSSON 1970). Chromatographie sur colonne, sur papier; chromatographie gazeuse. Chromatographie en phase vapeur (...) pour l'analyse des mélanges d'acides gras (Hist. gén. des sc., t. 3, vol. 2, 1964, p. 436).Rem. 1. Chromatographie entre comme second élément de compos. dans la formation de termes techn. Électrochromatographie (cf. électrophorèse). 2. On rencontre ds la docum. a) Le subst. masc. chromatogramme. Résultat visible et mesurable de l'analyse chromatographique (cf. H. PRIVAT DE GARILHE, Les Acides nucléiques, 1963, p. 45). b) Le subst. masc. chromatographe. Instrument servant à l'analyse chromatographique (cf. Hist. gén. des sc., t. 3, vol. 2, 1964, p. 431).Prononc. :[ ]. Étymol. et Hist. 1929 (Lar. 20e Suppl.). Composé de l'élément préf. chromat(o)- et de l'élément suff. -graphie (élément suff. -graphe, suff. -ie).DÉR. Chromatographique, adj. Relatif à la chromatographie. Analyse chromatographique (P. MORAND, Aux Confins de la vie, 1955, p. 89). — 1re attest. 1955 id.; de chromatographie, suff. -ique.chromatographie [kʀɔmatɔgʀafi] n. f.❖♦ Didact. Méthode d'analyse chimique par absorption sélective des constituants d'un mélange par une matière pulvérulente (les couches obtenues peuvent être diversement colorées). — REM. Le procédé lui-même date de 1906 (→ Chromatogramme).➪ tableau Vocabulaire de la chimie.❖DÉR. Chromatographique. — V. Chromatogramme.
]. Étymol. et Hist. 1929 (Lar. 20e Suppl.). Composé de l'élément préf. chromat(o)- et de l'élément suff. -graphie (élément suff. -graphe, suff. -ie).DÉR. Chromatographique, adj. Relatif à la chromatographie. Analyse chromatographique (P. MORAND, Aux Confins de la vie, 1955, p. 89). — 1re attest. 1955 id.; de chromatographie, suff. -ique.chromatographie [kʀɔmatɔgʀafi] n. f.❖♦ Didact. Méthode d'analyse chimique par absorption sélective des constituants d'un mélange par une matière pulvérulente (les couches obtenues peuvent être diversement colorées). — REM. Le procédé lui-même date de 1906 (→ Chromatogramme).➪ tableau Vocabulaire de la chimie.❖DÉR. Chromatographique. — V. Chromatogramme.
Encyclopédie Universelle. 2012.